
VOLUMEN: XI NÚMERO: 28
VOLUMEN: XI
NÚMERO: 28
ESTRÉS, DEPRESIÓN, INFLAMACIÓN y DOLOR.
Sánchez, P.T.
Sirera, R.
Peiró, G.
Palmero, F.
1. Contextualizar el Estrés y el Dolor.
El término “Estrés” se utiliza tan frecuentemente que resulta verdaderamente difícil comprender su significado. Su uso como sinónimo de tensión, ansiedad, nerviosismo, angustia, desazón... nos muestra que se maneja el término con ligereza para referirse a varios aspectos de un mismo concepto, cuando no para expresar conceptos diferentes. En las últimas décadas se ha aplicado indistintamente para designar a un estímulo, una respuesta, o una consecuencia. Se le ha denominado como fenómeno, estado o proceso, mostrando que no existe una visión única del estrés, ni un consenso en su definición como término ni como concepto psicológico, y ante todo que se habla desde modelos o perspectivas muy diferentes.
En la actualidad el modelo psicológico que aparece más consolidado en la literatura es el denominado interaccionista o integrador que englobaría tanto a los estímulos como a las respuestas y su interacción (Lazarus & Folkman, 1984), siendo su concepto central la existencia de una evaluación cognitiva de dificultad o incapacidad para responder a ciertas demandas sean estas externas o internas.
En el presente artículo proponemos utilizar explícitamente el concepto de estrés para significar el proceso psicobiológico global, entendiendo por tanto que nos referimos al conjunto de los procesos bioquímicos y neuropsicológicos que se dan en la persona que lo padece (Mateo, 2003).
Por otra parte, con el concepto “Dolor” queremos aprehender globalmente un fenómeno que no siendo uniforme (en realidad deberíamos utilizar el plural más que el singular), se presenta para el observador con una elevada complejidad (Munné, 2004). Se trata de un fenómeno generado a través de la interacción de nuestra estructura neuropsicológica y de su sustrato bioquímico, que adquiere su pleno significado personal cuando accede a nuestra conciencia (Simón, 2001). Solamente al hacerse consciente se transforma en experiencia cognitiva y emocional (Simón, 2002; Simón, 2003), que por muy habitual que sea para la humanidad, es percibida y sentida de forma totalmente peculiar por cada una de las personas que lo padecen.
El dolor – experiencia, como el sufrimiento, pertenecen por tanto en un inicio al ámbito de nuestra intimidad. Solamente cuando las compartimos abandonan definitivamente el ámbito de lo personal para convertirse en una experiencia relacional. Lo transmitimos según nuestra vivencia y cultura a través de las calificaciones y cualidades que le atribuimos: agudo, amargo, atroz, errático, fuerte…
Sin embargo cuando rememoramos la experiencia de nuestro dolor para transmitirlo, ha sido tamizado por el filtro de nuestra memoria de forma que comunicamos no la realidad misma, sino una memoria de esa realidad, una memoria que podríamos entender como enriquecida respecto a la original. El dolor como experiencia no es por tanto un concepto estático sino procesual, en continua revisión por nuestra conciencia, cuyo significado e implicaciones personales y sociales variará con el transcurso del tiempo, con nuestras vivencias e incluso con nuestro estado emocional.
La experiencia del dolor podría ser considerada siguiendo esta línea de pensamiento algo diferente al fenómeno del dolor, como la mente puede ser considerada algo diferente al cerebro. Lejos de representar un dualismo, esta postura sistémica (Kauffman, 2003; Bunge 1985) aboga por considerar que las propiedades emergentes, presentan una diferencia cualitativa fruto de la organización de la complejidad de los sistemas psiconeurobioquímicos. Por tanto, en este artículo, cuando citemos simplemente dolor será para referirnos a la experiencia de dolor.
2. Estrés.
Haciendo una breve sinopsis (ver la Figura 1) las vías clásicas del estrés involucran inicialmente al Hipotálamo que integra la información procedente de las vías sensoriales y viscerales, el Hipotálamo activaría dos rutas paralelas: el eje SAM (Simpático-Adreno-Medular) y el eje HPA (Hipotalámico-Pituitario-Adrenal) (Sirera et al, 2006).
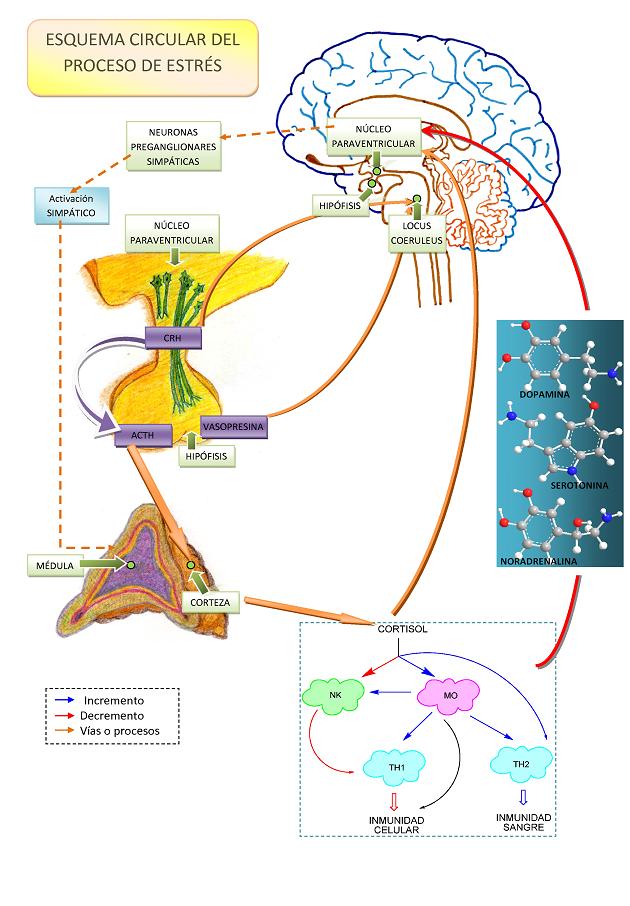
Figura 1
El eje SAM se inicia cuando las neuronas preganglionares simpáticas de la medula espinal reciben la información procedentes del hipotálamo activando la Rama Simpática e inhibiendo la Rama Parasimpática, esta activación genera cambios dirigidos a preparar al cuerpo para el esfuerzo físico sostenido y la toma de decisiones. La activación de las neuronas postganglionares simpáticas produce la liberación de Noradrenalina que es segregada a nivel de la médula suprarrenal y en estructuras cerebrales: hipotálamo, sistema límbico, hipocampo y córtex cerebral. Por otra parte las neuronas preganglionares simpáticas activan la médula de las glándulas adrenales liberando a la circulación Adrenalina y en menor medida Noradrenalina, generando un incremento de los niveles plasmáticos de glucosa y de ácidos grasos, también se incrementa la producción de tiroxina, mientras que se produce una disminución de los niveles de insulina, estrógenos y testosterona, e inhibición de la secreción de prolactina.
Paralelamente el Eje HPA se inicia a partir de la activación del Núcleo Paraventricular del Hipotálamo y tiene como objetivo mantener los parámetros de esfuerzo y atención. Las neuronas del núcleo paraventricular segregan a través de los vasos portales a la Adenohipófisis la Hormona Liberadora de Corticotropina (CRH). La CRH y otras hormonas relacionadas entran en el sistema circulatorio que une el hipotálamo con la pituitaria anterior, y activando la pituitaria, se libera corticotropina (ACTH) y en menor medida β-endorfina.
Por otra parte la activación de la Neurohipófisis por las neuronas magnocelulares del hipotálamo genera la segregación de Vasopresina y Oxitocina que potenciaran el efecto de la CRH. Una vez secretada la Corticotropina en el flujo sanguineo estimula la producción y liberación de Glucocorticoides (cortisol y corticosterona) y mineralocorticoides por parte de las Glandulas Adrenales. Los efectos de altos niveles de glucorticoides (especialmente el cortisol) a medio / largo plazo en la salud son realmente perjudiciales: aumento en la presión sanguínea, daño en el tejido muscular... por lo que existen circuitos de retroalimentación que intentan mantener sus niveles sanguíneos dentro de unos parámetros definidos.
La activación del sistema Inmune (Hansen-Grant et al, 1998; Reiche et al, 2004) por parte de los ejes SAM y HPA se produce a través de dos mecanismos diferentes, por un lado la unión de las hormonas a sus receptores cognatos y por otro indirectamente a través de la desregulación del equilibrio que tiene que imperar en la producción de citocinas proinflamatorias (Sirera et al, 2006b; Maes et al, 1998). Las citocinas proinflamatorias son mediadores solubles que fomentan y median los procesos inflamatorios, destacan: la interleucina 1 (IL-1) interviene en la regulación del proceso inmune y de la inflamación; la interleucina 6 (IL-6) que además sirve de enlace entre el Sistema endocrino y el Sistema Inmune; y el Factor de Necrosis Tumoral (TNF) que posee la capacidad de destruir ciertas líneas celulares e inicia la cascada de citocinas proinflamatorias y otros mediadores. Existen datos de que además de con el sistema endocrino las citocinas IL-1, IL-6 y TNFα interactúan con los sistemas Noradrenérgico, Serotoninérgico, y Dopaminérgico (Kronfol & Remick, 2000).
3. Inflamación.
La inflamación es un proceso bioquímico que puede ser originada por numerosos factores endógenos o exógenos, de hecho cualquier fenómeno inmunológico capaz de afectar a la estabilidad del sistema puede ser considerado como un estresor y al proceso denominarlo como estrés inflamatorio. El Sistema Inmune tiene como función el reconocer y destruir tanto a patógenos externos como internos. Para realizarlo posee dos sistemas intercomunicados: la inmunidad innata o inespecífica y la inmunidad adquirida o específica que se suele subdividir en dos grupos complementarios: inmunidad celular e inmunidad humoral. Son las células del sistema inespecífico (neutrófilos, macrófagos y dendríticas) las que inician la respuesta inmune a través de la fagocitosis y la inflamación. La inmunidad inespecífica induce la inmunidad específica, y como resultado se genera una respuesta especializada y podríamos decir con memoria. La intensidad, duración y características peculiares de las inflamaciones dependerán del área afectada, del estado previo y de la causa que la provoca.
La inflamación crónica se produce como resultado de la presencia durante un tiempo prolongado de algún agente infeccioso, antígeno o por causa de un trastorno del sistema inmunológico. Cada vez disponemos de más datos que sugieren que la inflamación puede contribuir al desarrollo de enfermedades como el Alzheimer (Tan et al, 2007), el cáncer (Pikarsky et al, 2004), la ateroesclerosis (Piñon & Kaski, 2006), y la diabetes (Rosado y Mendoza, 2007), además de en aquellas donde los procesos inflamatorios son la misma base de la enfermedad como el Crohn o la artritis reumatoide. La inflamación crónica se caracteriza por la formación de tejido fibroso y que el infiltrado celular está compuesto sobre todo por macrófagos, linfocitos y células plasmáticas. Entre los mediadores de la inflamación debemos destacar el papel de las citocinas especialmente la IL-1ß y la TNFα capaces de activar numerosas cascadas humorales de mediadores que perpetúan la activación del sistema. Las citocinas (ver Tabla 1) forman un grupo importante de proteínas que actúan como mediadores de la comunicación entre células vivas. Para entender el papel de las citocinas es necesario comprender sus mecanismos de acción que lo son tanto a nivel local como general, se han postulado varios no excluyentes: transporte pasivo a la zona circunventricular, unión al endotelio vascular y posterior liberación por parte de otros agentes (prostaglandinas, oxido nítrico) al interior del cerebro, transporte activo a través de la barrera hematoencefálica y activación periférica de las terminaciones nerviosas donde se ha producido la liberación (Watkins el al, 1995).
TABLA 1. CUADRO DE CITOCINAS.
|
CITOCINAS |
Fuente |
Acción |
|
IL-1: IL-1α, IL-1β, IL-1RA. |
Macrófagos, Monocitos y Células Dendríticas |
Proinflamatoria. Activa las células T y estimula el crecimiento medular (Dinarello, 2000 y2005; Arend & Gabay, 2000) |
|
IL-2. |
|
Promueve la proliferación de células T. Interviene en la respuesta inflamatoria, induce la liberación de IL-1, TNFα y TNFβ (Smith, 2006) |
|
IL–4 |
Linfocitos Th0 activados y los Mastocitos. |
Promueve la proliferación de células T y la activación, proliferación y diferenciación de los Linfocitos B.(Kelly-Welch et al, 2003) |
|
IL-5 |
Linfocitos Th2 y los Mastocitos. |
Activación, proliferación y diferenciación de Linfocitos B, y principal factor regulador de la eosinofilia. (Schwenger et al, 2001) |
|
IL-6 |
Macrófagos activados, Monocitos, Fibroblastos y las Células del endotelio vascular. |
Principal regulador de la respuesta inmunológica y de reacciones de fase aguda, interviene también en la hematopoyesis. Tiene propiedades proinflamatorias y antiinflamatorias, predominando la acción antiinflamatoria. (Spranger et al, 2003) |
|
IL-8 |
Monocitos. |
Moviliza y activa a los Neutrófilos, estimulando la fagocitosis. Es también angiogénico .(Xie, 2001) |
|
IL-10 |
Linfocitos Th2. |
Tiene efecto antiinflamatorio. Inhibe la expresión de IFN-g e IL-2 por parte de los LTh1, y la síntesis de IL-1, IL-6 y TNF-a por parte de los Macrófagos. Es también co-estimulador del crecimiento de varias células hematopoyéticas, incluyendo Linfocitos B y T. (Perez & Kaski, 2002) |
|
IL-12 |
Linfocitos B. |
Induce la síntesis de IFN-g e IL-2 por los LTh1 e inhibe la producción de IL-4, IL-5 e IL-10 por los LTh2. Además activa las células NK y aumenta la expresión de receptores para la IL-18. (Mannon et al, 2004) |
|
IL-13 |
Linfocitos Th2 y Mastocitos. |
Estimula e incrementa la producción de citocinas por los LTh2, e inhibe las citocinas proinflamatorias sintetizadas por los LTh1 y Macrófagos, y factores quimiotácticos secretados por Monocitos y Linfocitos B. .(Kelly-Welch et al, 2003) |
|
IL-16 |
Linfocitos T8 activados. |
Es proinflamatoria. También denominada factor quimioatrayente de Linfocitos, aumenta la expresión de receptores para la IL-2 en Linfocitos B y T, células NK, Macrófagos y Monocitos. (Cruikshank et al, 2000) |
|
IL-18 |
Macrófagos activados. |
Acción similar a la IL-12 con la que actúa sinérgicamente. Es la mayor inductora de la producción de IFN-g. (Dinarello, 2007; Kim et al, 2007) |
|
TNFα |
Macrófagos, Monocitos y los Linfocitos T. |
Tiene efecto anti-tumoral y actúa como mediador en el desarrollo del shock séptico y de la caquexia en enfermedades crónicas. Además activa potentemente a los Macrófagos, estimula la producción de citocinas por muchos tipos celulares e incrementa la permeabilidad vascular. (Kronfol & Remick, 2000) |
|
TNFβ |
Linfocitos T activados. |
Tiene actividad citotóxica sobre algunos tipos tumorales, mediado solo por necrosis hemorrágica. |
Las IL-1ß y la TNFα están presentes desde el inicio en la secuencia de actividades que buscan la liberación y utilización de la glucosa para la reparación del tejido o la elevación de la temperatura corporal, induciendo la producción de una segunda ola de citocinas, IL-1, IL-6, IL-8 y el Macrophage Chemotactic Protein, derivados del ácido araquidónico o eicosanoides, factor activador de las plaquetas (PAF), la liberación de radicales libres y la producción de óxido nítrico (NO), también citocinas antiinflamatorias (IL-4 e IL-10) y la liberación de otras sustancias mediadoras de la inflamación (Nonaka, 2001) como las bradiquininas, histamina… La IL-1 y la IL-6 actúan sobre el eje HPA (Chrousos, 1995) aumentando la secreción de ACTH y de cortisol. La función reguladora-supresora de la respuesta inmune dependerá entonces del balance entre la síntesis de distintas citocinas con acciones diversas. Si la inflamación se prolonga se activarán además otros sistemas: el Endocrino, el Noradrenérgico, el Serotoninérgico y el Dopaminérgico (Kronfol & Remick, 2000).
El estrés inflamatorio es inmunomodulador, en este sentido se ha hipotetizado la existencia de un posible patrón de inhibición de la respuesta inmune celular (Th1) mientras se incrementa la inmunidad humoral (Th2) o viceversa (Singh et al, 1999; Agarwal & Marshall, 1998). Las Th1 producen (IFN-γ, TNF-β) activadores de macrófagos, en contrate las Th2 producen IL-4, IL-5, IL-10, y IL-13 activadoras de la respuesta de los anticuerpos, también podrían existir otros modelos. Sin embargo los resultados observados en procesos clínicos como el Lupus eritomatoso sistémico (Chang et al, 2002) o en cáncer de próstata (Filella et al, 2000) han sido calificados de ambiguos. Acumulamos datos que sugieren que las citoquinas proinflamatorias son capaces de: activar tanto el Eje HPA como el sistema Locus Coeruleus – Norepinefrina del Eje SAM. Estimular las concentraciones plasmáticas de glucocorticoides, alterando la actividad de las neuronas noradrenérgicas hipotalámicas, disminuyendo la norepinefrina en el bazo. Los glucocorticoides por su parte inhiben la secreción de IL-2, IFNγ e IL-12 mientras incrementan la secreción de IL-4 e IL-10. La inflamación crónica o intermitente que se genera por la presencia de un foco infeccioso puede así asociarse al estrés, siendo capaz de iniciar la cascada de procesos bioquímicos que la hacen formalmente indistinguible del propio proceso psicobiológico.
4. Depresión.
La Depresión es un Síndrome o Trastorno del estado de ánimo caracterizado por la presencia criterial de un conjunto de síntomas: Tristeza, Anhedonia, Astenia o Lasitud, disminución de la atención y concentración, perdida de la autoconfianza, Pesimismo, ideación de muerte o suicidio, insomnio, anorexia … En el modelo clásico surgido en la década de 1960 se implicaba en el desarrollo del síndrome básicamente al sistema Serotoninérgico y al Sistema Noradrenérgico (Ressler & Nemeroff, 2000; Mongeau et al, 1997; Nemeroff. 2002). Sin embargo, en los últimos años se han estudiado en profundidad diversos procesos bioquímicos capaces de generar cambios en el estado de ánimo. Entre ellos podemos destacar la inflamación, la isquemia, la necrosis, la apoptosis… De todos ellos probablemente sea el de la inflamación el más estudiado y en el que disponemos de más datos que sugieren la relación entre el curso de la depresión y el de la inflamación (Licinio & Wong, 1999).
El puente teórico para relacionar la inflamación y la depresión, así como el estrés (tanto psicológico como físico), está conformado por las citocinas (Connor & Leonard, 1998 y de manera más específica por las interleucinas especialmente las proinflamatorias. Las interleucinas proinflamatorias interactúan con los Sistemas Endocrino, Noradrenérgico, Serotoninérgico y Dopaminérgico (Kronfol & Remick, 2000). En los modelos bidireccionales más actuales se considera que tanto el estrés, como la depresión y la inflamación son capaces de activar y modificar el equilibrio de las citocinas y viceversa. Como ejemplo, un incremento de las interleucinas proinflamatorias (IL-1, IL-6, TNFα) independientemente de su origen podemos relacionarlo con incrementos en la Noradrenalina, Serotonina, Dopamina, Cortisol, Hormona Liberadora de Corticotropina CRH, y Corticotropina ACTH, junto con un descenso de la Hormona Liberadora de Gonadotropina GnRH y de la actividad de las Natural Killer cells (Kronfol & Remick, 2000).
Las evidencias más fuertes del rol de las citocinas en la depresión provienen de la observación clínica de animales (Felger et al, 2007) y pacientes en tratamiento con interferones (Asnis & La Garza, 2005). Así la administración de interferón-α (Gleason & Yates, 1999) (en Hepatitis C o Melanoma), de interferon-β (en Esclerosis Múltiple), interferon-γ (Sarcoma de Kaposi o en la Enfermedad Granulomatosa Crónica) o de la interleucina-2 (en Cáncer Metastásicos), se asocian con cambios afectivos y comportamentales que incluyen el desarrollo de episodios depresivos. Otras evidencias que sugieren el rol del sistema inmune en el desarrollo y la consolidación de la depresión incluyen las observaciones de que los pacientes deprimidos muestran: elevados niveles de IL-6 (Chrousos, 1995; Maes et al, 1993); elevados niveles de reactantes de fase aguda y marcadores de activación de las células inmunes, así como función inmune alterada (Maes et al, 1993). Las citocinas parecen ejercer un efecto depresivo, directamente por medio de la activación la hormona liberadora de corticotropina, o indirectamente provocando resistencia de los receptores de glucorticoides, lo que causa hiperactividad del eje hipotálamo-hipofisiario-suprarrenal, debido a inhibición del mecanismo de retroalimentación normal. La administración intracraneal de citocinas proinflamatorias provoca los mismos efectos de enfermedad que su administración sistémica. Por lo tanto las citocinas proinflamatorias tienen dos lugares donde ejercer su acción claramente distintos, en el mismo lugar de la inflamación y en el SNC.
El estrés ya sea físico o psicológico juega un rol importante en el desencadenamiento y evolución de los trastornos depresivos. Además, en la depresión se ha mostrado la existencia de un perfil bioquímico a nivel endocrino e inmunológico similar al observado en el estrés. Los glucocorticoides (Burke et al, 2005) se encuentran entre los principales mediadores de los efectos inmunosupresores generados por estresores. Entre los efectos descritos en humanos encontramos linfocitopenia (por ejemplo tras enviudar), monocitopenia, y neutropenia. También los glucocorticoides parecen estar implicados en la reducción de la producción de determinadas citocinas (IL-1, IL-2, IL-6, IL-8, TNF) o en los incrementos de otras como la IgA, IgE, IgG o la IgM. Además los glucocorticoides ejercen también su acción en el cerebro atravesando la barrera hematoencefálica. En este sentido se han encontrado receptores de mineralcorticoides entre otras localizaciones en las neuronas piramidales y granulares del hipocampo, el núcleo olfativo, la amígdala, el estriado y el septum. Los receptores de glucocorticoide están presentes en una alta proporción en el hipocampo y en el hipotálamo, además los encontramos en la corteza frontal, parietal y entorrinal entre otros. Por consiguiente los glucocorticoides pueden modular diversos procesos en el sistema nervioso central.
En los últimos años, el sistema dopaminérgico está adquiriendo un peso específico (Dunlop & Nemeroff, 2007) en la fisiopatología de la depresión. La función excitatoria de la Dopamina en las neuronas del Núcleo Paraventricular, donde la activación de los receptores dopaminérgicos D1 y D2 estimula el eje HPA y propicia la secreción de corticosterona, estableciendo un sistema de retroalimentación positiva. Y también las relaciones que se estas encontrando entre las neuronas con receptores D2 y las proteínas PAR-4 (Gurumurthy et al, 2005; Park et al, 2005; Mattson & Gleichmann, 2005).
5. Dolor.
La relación entre el Estrés y el Dolor no es un tema novedoso pues viene siendo discutida desde hace décadas lo que nos ha permitido acumular numerosos datos y experiencias que nos muestran la existencia de una relación cuasi circular donde los niveles del estrés y dolor se retroalimentan al utilizar vías comunes. Sintéticamente las líneas de trabajo (Keefe et al, 2004) que más se manejan en la actualidad parecer ser la analgesia inducida por estrés, el estrés como amplificador del dolor y el dolor como estresor.
Respecto a la analgesia inducida por estrés, desde siempre se ha observado como personas sometidas a una situación de elevado estrés agudo (accidente, herida, …) reaccionaban inmediatamente sin sentir aparentemente dolor a pesar de la gravedad de sus lesiones. En los últimos años parece haberse reactivado el interés por descubrir los procesos bioquímicos implicados en esta analgesia, estudiando los mecanismos de acción de los endocannabinoides (Hohmmann et al, 2005; Connell et al, 2006), de la noradrenalina (Delaney et al, 2007), y de la interleucina 1 (IL-1) (Wolf et al, 2007).
El estrés se ha señalado también como un amplificador del dolor, especialmente entre los pacientes que presentan una mayor hipersensibilidad y activación (Ursin & Eriksen, 2001), o con un umbral incrementado de reactividad frente a los estimulos (Cacioppo et al, 1998; Montoya et al, 2005). Una posibilidad complementaria es valorar el propio dolor crónico como un potente estímulo estresor, generador de sintomatología y de comorbilidad psicopatológica (Dersh et al, 2002; Greenberg & Burns, 2003). Los procesos bioquímicos implicados en esta relación son numerosos aunque destacan las hormonas inducidas por emociones negativas (Chrousos & Gold, 1998), catecolaminas (adrenalina y noradrenalina), la adenocorticotropa (ACTH), el cortisol (Melzack, 2000), la hormona del crecimiento y la prolactina, capaces de inducir cambios cualitativos y cuantitativos en el sistema inmulógico. Fundamentalmente el cambio se produce incrementando la producción de citocinas (Zhou et al, 1993), y esos incrementos se pueden asociar tanto a los cambios emocionales como a los comportamentales. Igualmente se ha observado la liberación de citocinas durante el estrés quirúrgico a pesar de la utilización de potentes anestésicos (McBride, 1996; Moore et al, 1994).
Otra línea de relación puede establecerse entre los procesos inflamatorios y la amplificación de la sensibilidad al dolor. Aunque existen diferentes vías posibles, en las lesiones traumáticas y quirúrgicas se han observado cambios en el sistema inmunológico (Van Dijk et al, 1999; Krieglstein, 1999; Oberholzer & Moldawer, 2000) que incluyen un exceso de producción de citocinas proinflamatorias y/o antiinflamatorias. Así se encuentran datos de disminución de IL-5 tras hemorragias masivas (Slifka & Whitton, 2000), y en pacientes politraumatizados un incremento de células T y de factores solubles inmunosupesores (Suffredini & Openheim, 1999). Recordemos que las citocinas proinflamatorias aumentan la actividad en las rutas nociceptivas y pueden causar sensibilización al activar la liberación de otras citocinas y mediadores clásicos de la hiperalgesia, como prostanoides, aminas simpatomi-méticas, endotelina o glutamato. Esta sensibilización neuronal ocurre tanto centralmente como en la periferia. Las citocinas podrían desencadenar el dolor promoviendo la inflamación, aún en la ausencia de lesión o daño. Por ello se impone profundizar en nuestro conocimiento sobre el papel preciso que las citocinas desempeñan en la producción y en el mantenimiento del dolor. Recientemente se ha propuesto (Ikeda et al, 2006) el incremento de la sensibilidad al dolor mediante cambios en la excitabilidad de la médula espinal por la elevación de los niveles de iones de calcio, lo cual alteraría el procesamiento medular de la propia sensación de dolor. En la medula espinal también la glía (Watkins & Maier, 2003) puede jugar un papel importante, dado que libera citocinas proinflamatorias que junto a las antiinflamatorias median y modulan el mensaje del dolor.
6. A modo de conclusión.
La complejidad de la relación entre cada uno de los procesos psicobiológicos expuestos, debemos valorarlo como un incentivo para generar cada vez más conocimientos y abrir nuevos caminos para la investigación de los mecanismos moleculares que lo sustentan. Sin embargo y en la misma medida que avanzamos en las vías y mecanismos moleculares, también creemos que es muy importante reflexionar (Le Breton, 1999) junto a otras disciplinas, sobre los motivos psicosociales por los cuales toda esta progresión y acumulación de conocimientos no está siendo útil para evitar el sufrimiento generado por un dolor, que en demasiados pacientes parece refractario a todos los tratamientos disponibles (Turk, 2002). O tal vez, debamos reflexionar sobre la relación existente entre el sufrimiento y el dolor, cuando empezamos a obtener que el sufrimiento ya supera al propio dolor (Bushnell et al, 1999) o que es el sufrimiento el que incrementa la sensibilidad frente a él (Rode et al, 2001).
7. Bibliografia.
Agarwal SK, Marshall GDJ. (1998) Glucocorticoid induced type 1 type 2 cytokine alterations in humans: a model for stress-related immune disfunctions. J Interferon Cytokine Res 18:1059-1068.
Arend W, Gabay C. (2000) Physiologic role of interleukin-1 receptor antagonist. Arthritis Res. ;2:245-248.
Asnis GM, La Garza R. (2005) Interferon-induced depression: strategies in treatment. Prog Neuropsychopharmacol Biol Psychiatry; 29:808-818.
Bunge M. (1985) El problema mente-cerebro. Un enfoque psicobiológico. Ed. Tecnos: Madrid.
Burke HM, Davis MC, Otte C, et al. (2005) Depression and cortisol responses to psychological stress: a meta-analysis. Psychoneuroendrocrinology; 30: 846 – 856.
Bushnell MC, Duncan GH, Hofbauer RK, et al. (1999) Pain Perception: Is there a role for primary somatosensory cortex? Proc Natl Acad Sci USA ; 96: 7705-7709.
Cacioppo JT, Berntson GG, Malarkey WB, et al. (1998) Autonomic, neuroendocrine, and inmune responses to psychological stress: the reactivity hipótesis. Ann N Y Acad Sci ; 840: 664 – 673.
Chang DM, Su WL, Chu SJ. The expression and significance of intracellular T helper cytokines in systemic lupus erythematosus. Immunol Invest 2002;31:1-12.
Chrousos GP. (1995) The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med 332:1351-62.
Chrousos GP, Gold PW. (1998) A healthy body in a healthy mind--and vice versa--the damaging power of “uncontrollable” stress. J Clin Endocrinol Metab ; 83:1842-5.
Connell K, Bolton N, Olsen D, et al. (2006) Role of the basolateral nucleus of the amygdala in endocannabinoid-mediated stress-induced analgesia. Neurosci Lett; 397: 180-184.
Connor TL, Leonard BE. (1998) Depression, stress, and immunological activation. The role of cytokines in depresive disorders. Life Science; 7:583-606.
Cruikshank WW, Kornfeld H, Center DM. (2000) Interleukin-16. J Leukoc Biol ; 67: 757 – 766.
Delaney AJ, Crane JW, Sah P. (2007) Noradrenaline modulates transmission at a central synapse by a presynaptic mechanism. Neuron; 56:880-892.
Dersh J, Polantin PB, Gatchel RJ. (2002) Chronic pain and psychopathology: research finding and theoretical considerations. Psychosom Med ; 64: 773 – 786.
Dinarello CA. (2000) The role of the interleukin-1 receptor antagonist in blocking inflammation mediated by interleukin-1. N. Engl J Med. ;343:732-734.
Dinarello CA. (2005) Blocking IL-1 in systemic inflammation. J Exp Med. ;201: 1355–1359.
Dinarello CA. (2007) Interleukin-18 and the pathogenesis of inflammatory diseases. Semin Nephrol. ;27:98–114.
Dunlop BW, Nemeroff CB. (2007) The role of dopamine in the pathophysiology of depression. Arch Gen Psychiatry; 64: 327 - 337.
Felger JC, Alagbe O, Hu F, et al. (2007) Effects of interferon-alpha on rhesus monkeys: a nonhuman primate model of cytokine-induced depression. Biol Psychiatry; 62: 1324 - 1333.
Filella X, Alcover J, Zarco MA, et al. Analysis of type T1 and T2 cytokines in patients with prostate cancer. Prostate 2000;44:271-274
Gleason OC, Yates WR. (1999) Five cases of interferon alpha-induced depresion treated with antidepresant therapy. Psychosomatics; 40: 510-512.
Greenberg J, Burns JW. (2003) Pain anxiety among chronic pain patients: specific phobia or manifestation on anxiety sensitivity?. Behav Res Ther ; 41: 223 – 240.
Gurumurthy S, Goswami A, Vasudevan Km, et al. (2005) Phosphorylation of Par-4 by protein kinase A is critical for apoptosis. Moll Cel Biol; 25: 1146 – 1161.
Hansen-Grant SM, Pariante CM, Kalin NH, et al. (1998) Neuroendocrine and immune system pathology in psychiatric disease. In: Schatzberg AF, Nemeroff CB, eds. Textbook of Psychopharmacology, 2nd ed. Washington, DC: American Psychiatric Press:171-194.
Hohmmann AG, Suplita RL, Bolton NM, et al. (2005) An endocannabinoid mechanism for stress-induced analgesia. Nature; 435:1108-1112.
Ikeda H, Stark J, Fischer H, et al. (2006) Synaptic Amplifier of Inflammatory Pain in the Spinal Dorsal Horn, Science ; 312: 1659 – 1662.
Kauffman SA. (2003) Investigaciones: complejidad, autoorganización y nuevas leyes para una biología general. Tusquets. Barcelona.
Keefe FJ, Rumble ME, Scipio CD, et al. (2004) Psychological aspects of persistent pain: current state of the science. J Pain; 5: 195 – 211.
Kelly-Welch AE, Hanson EM, Boothby MR, et al. (2003) Interleukin-4 and Interleukin-13 signaling connections maps. Science; 300: 1527 – 1528.
Kim HJ, Kang ES, Kim DJ, et al. (2007) Effects of rosiglitazone and metformin on inflammatory markers and adipokines: decrease in interleukin-18 is an independent factor for the improvement of homeostasis model assessment-beta in type 2 diabetes mellitus. Clin Endocrinol (Oxf). ;66:282–289.
Krieglstein K. (1999) Synergy effects of cytokines lead to development of neurotrophic funtions. Ann Anat ; 181: 423-427.
Kronfol z, Remick DG. (2000) Cytokines an the brain: implications for clinical psychiatry. Am J Psychiatry 157: 683 – 694.
Lazarus RS, Folkman S. (1984) Stress, Appraisal, and Coping. New York. Springer.
Le Breton D. (1999) Antropología del dolor. Seix Barral. Barcelona.
Licinio J, Wong ML. (1999) The role of inflammatory mediators in the biology of major depresion: central nervous system cytokines modulate the biological substrate of depresive symptoms, regulate stress-responsive systems, and contribute to neurotoxicity and neurodegeneracion. Mol Psychiatry; 4:317 – 327.
Maes M, Scharpe S, Meltzer HY, et al. (1993) Relationship between interleukin-6 activity, acute phase proteins, and the function of the hypothalamic-pituitaryadrenal axis in severe depression. Psychiatry Res;49:11-27.
Maes M, Song C, Lin A, et al. (1998) The effects of psychological stress on humans incresased production of pro-antinflammatory cytokines and aTH1-like response in stress-induced anxiety. Cytokine 10:313-318.
Mannon PJ, Fuss IJ, Mayer L, et al. (2004) Anti–Interleukin-12 Antibody for Active Crohn's Disease. N Engl J Med ; 351: 2069-2079.
Mateo MA. (2003) Notas sobre la complejidad en psicología. Anales de Psicología 19: 315 – 326.
Mattson MP, Gleichmann M. (2005) The neuronal death protein Par-4 mediates dopaminergic synaptic plasticity. Mol Interv 2005; 5:278 - 281.
McBride WT. (1996) Inmunomodulation: an important concept in modern anestesia. Anaesthesia ; 51: 465-473.
Melzack R. (2000) Del umbral a la neuromatriz. Rev Soc Esp Dolor ; 7: 149-156.
Mongeau R, Blier P, De Montigny C. (1997) The serotonergic and noradrenergic systems of the hippocampus: Their interactions and the effects of antidepressant treatments. Brain Res Rev; 23: 145-195.
Montoya P, Sitges C, García-Herrera M, et al. (2005) Abnormal affective modulation of somatosensory brain proccesing among patients with fibromyalgia. Psychosom Med ; 67: 957 – 963.
Moore CM, Desboroug JP, Powell H, et al. (1994) Effects of extradural anaesthesia in interleukin-G and acute phase response to surgery. Br J Anaesth ; 72: 272-279.
Munné F. (2004) El retorno de la complejidad y la nueva imagen del ser humano: hacia una Psicología Compleja. Interamerican J Psychol 38: 23 – 31.
Nemeroff CB. (2002) Recent Advances in Neurobiology of Depression. Psychopharmacol Bull; 32: 6 – 23.
Nonaka M. (2001) Evolution of the complement system. Curr Opin Immunol 13:69 -73.
Oberholzer C, Moldawer LL. (2000) Cytokine signaling-regulation of the immune response in normal and critically ill states. Crit Car Med ; 28: 4.
Park M, Nguyen A, Fischer M, et al. (2005) Par-4 Links Dopamine Signaling and Depression. Cell; 122: 275 – 287.
Perez R, Kaski JC. (2002) Interleucina 10 y enfermedad coronaria Rev Esp Cardiol 2002; 55: 738 – 750.
Piñón P, Kaski JC. (2006) Inflamación, aterosclerosis y riesgo cardiovascular: PAPP-A, Lp-PLA2 y cistatina C. ¿Nuevas aportaciones o información redundante? Rev Esp Cardiol 59: 247 – 258.
Pikarsky E, Porat RM, Stein I, et al. (2004) NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 431: 461-466.
Reiche EM, Nunes SO, Morimoto HK. (2004) Stress, depression, the immune system, and cancer. Lancet Oncol 5: 617-25.
Ressler KJ, Nemeroff CB. (2000) Role of serotoninergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depress Anxiety; 12: 2 - 19.
Rode S, Salkovskis PM, Jack T. (2001) An experimental study of attention, labeling and memory in people suffering from chronic pain. Pain ; 94: 193-203.
Rosado J, Mendoza VM. (2007) Mini Revisión: Inflamación Crónica y estrés oxidativo en la Diabetes Melitus. Bioquimia 32: 58 – 69.
Schwenger GT, Fournier R, Kok CC , et al (2001). GATA-3 Has Dual Regulatory Functions in Human Interleukin-5 Transcription. J. Biol. Chem. ; 276: 48502- 48509.
Simón VM. (2001) El ego, la conciencia y las emociones: un modelo interactivo. Psicothema 13: 205 – 213.
Simón VM. (2002) Las trampas de la imaginación. Psicothema 14: 643 – 650.
Simón VM. (2003) La deuda emocional. Psicothema 15: 328 – 334.
Singh VK, Mehrotra S, Agarwal SS. (1999) The paradigm of Th1 and Th2 cytokines: its relevance to autoimmunity and allergy. Immunol Res 20: 147-161.
Sirera R, Sánchez PT, Camps C. (2006) Inmunología, estrés, depresión y cáncer. Psicooncologia 3: 35 – 48.
Sirera R. González A. Camps C. (2006b) Papel del Sistema Inmunológico en el estado de ánimo. En Camps C y Sánchez PT Eds. Comunicación en Oncología. Seom.
Slifka MK, Whitton JL. (2000) Clinical implications of dysregulated cytokine production. J Mol Med ; 78: 74-80.
Smith KA. (2006) The structure of IL2 bound to the three chains of the IL2 receptor and how signaling occurs. Med Immunol ; 5: 3.
Spranger J, Kroke A, Mohlig M, et al. (2003) Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. ;52:812–817.
Suffredini AF, Openheim JJ.(1999) New insights into the biology of the acute phase response. J Clin Immunol ; 4: 203-209.
Tan ZS, Beiser AS, Vasan RS, et al. (2007) Inflammatory markers and the risk of Alzheimer disease: The Framingham Study. Neurology 68: 1902-1908.
Turk DC. (2002) Clinical effectiveness and cost effectiveness of treatments for chronic pain patients. Clin J Pain ; 18: 355-365.
Ursin H, Eriksen H. (2001) Sensitization, subjective health complaints and sustained arousal. Ann N Y Acad Sci ; 933: 119-129.
Van Dijk WC, Verbrugh HA, Van Rijswijk RE, et al. (1999) Neutrophyl function, serum opsonic activity, and delayed hypersensitivity in surgical patients. Surgery; 92: 21-29.
Watkins LR, Maier SF, Goehler LE. (1995) Cytokine-to-brain communication: a review & analysis of alternative mechanisms. Life Sci 57:1011-26.
Watkins LR, Maier SF. (2003) Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov ; 2: 973 -985.
Wolf G, Yirmina R, Kreisel T, et al. (2007) Interleukin-1 signaling modulates stress-induced analgesia. Brain Behav Inmun; 21: 652 – 659.
Xie K. (2001) Interleukin-8 and human cáncer biology. Cytokine Growth Factor Rev; 12: 375 – 391.
Zhou D, Kusnecov AW, Shurin MR, et al. (1993) Exposure to physical and psychological stressors elevates plasma interleukin 6: relationship to the activation of the hypothalamic-pituitary-adrenal axis. Endocrinology; 133: 2523-2530.